Our products help renal care and nephrology technicians keep dialysis machines functioning properly and allow testing and calibration for quality control of all types of delivery systems and water purification systems. Our user-friendly, time-saving meters and NIST-traceable solutions ensure accurate measurements to support safe, optimal dialysis therapy.
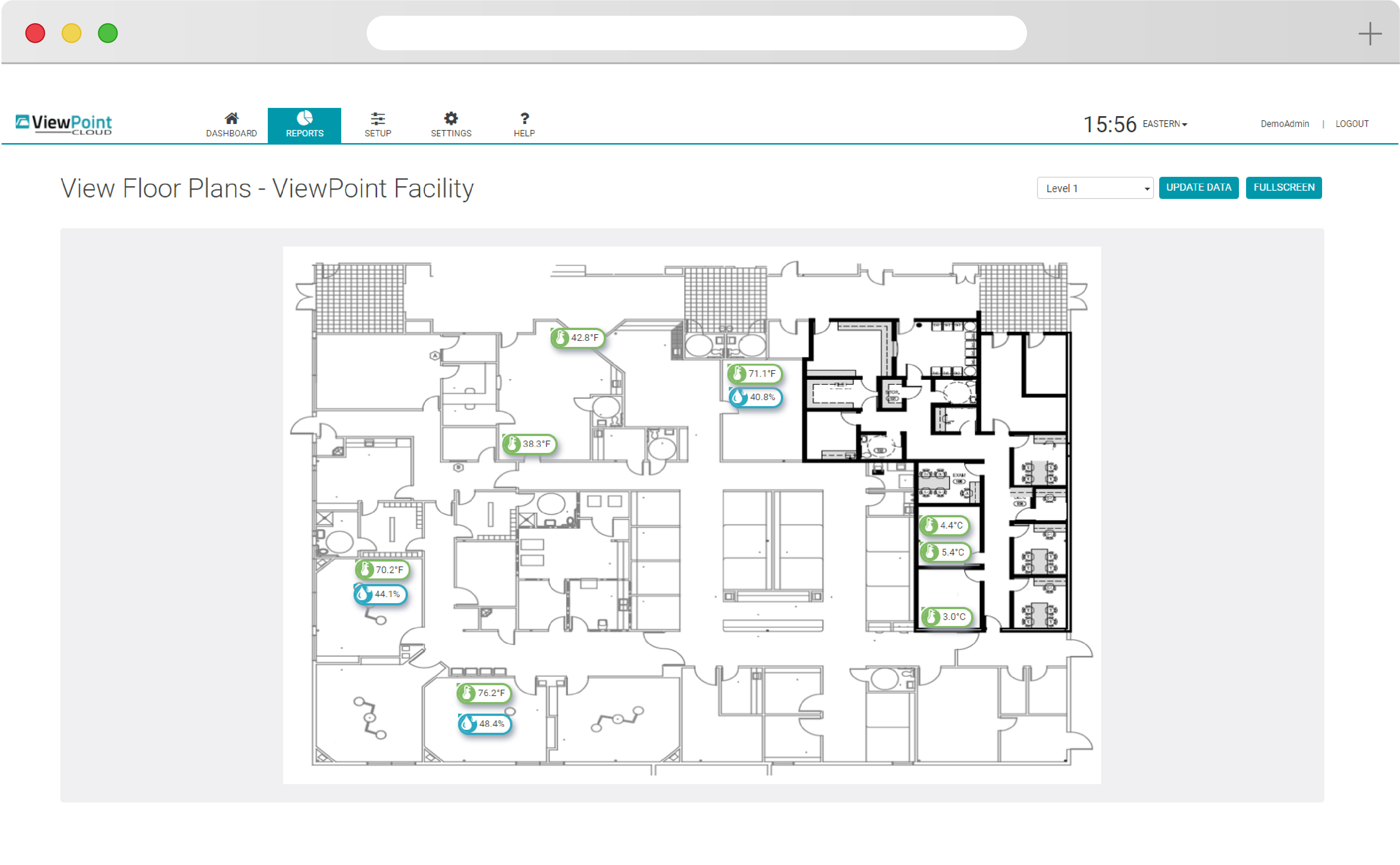
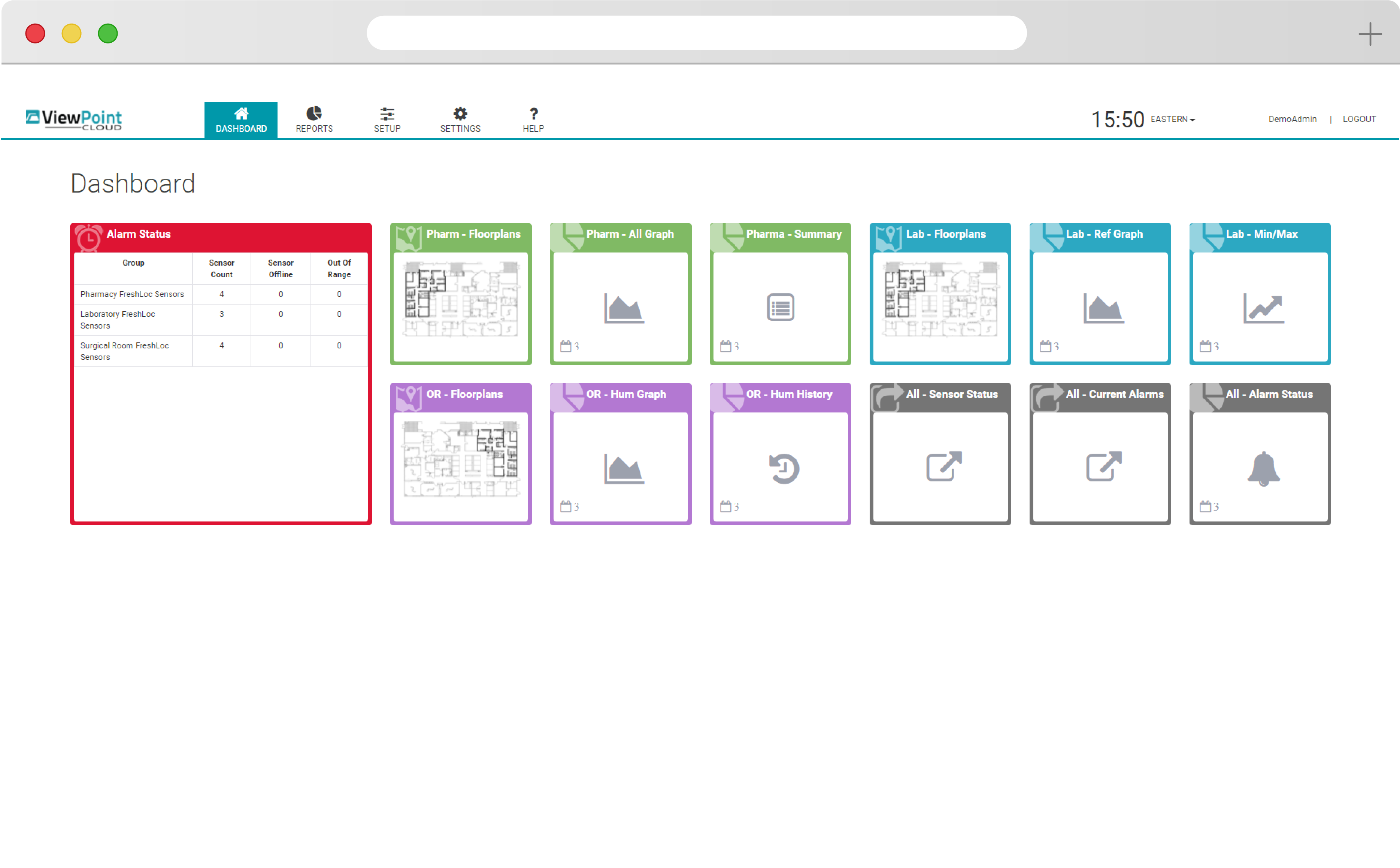
See your entire scope of oversight immediately with ViewPoint Cloud’s simple floorplan and overlay features. Build from an existing network, scale up with new hardware, or add new requirements and parameters to meet your evolving needs.
Easy
ViewPoint Cloud is easy to use, with intuitive drag-and-drop functionality and simple navigation that puts powerful insight at your fingertips. Quickly discern changes and set alerts that allow your team to react quickly to protect your products.
Our commitment to precision and ease of use make our products a top choice around the world for testing and calibrating all types of delivery and water purification systems.
Accurate
Our highly accurate digital dialysate meters are designed to deliver precise readings quickly. Easy field replaceable modules ensure ongoing calibration for greater reliability.
Trusted
Our digital dialysate meters are 510(K)-approved, test for greater ranges, are easier to use, and are more rugged to fit clinical environments than others on the market.
Comprehensive
We offer meters, NIST-traceable solutions, and ancillary products that make it easy to organize and optimize renal care quality control programs.
Renal Care Insights
Explore the latest in dialysis meter technology, trends, and insights for optimal renal care management.